
�������: 1-15 ���鵽����ѧ �������ؼ�¼54�� . ��ѯʱ��(0.165 ��)

�й���ѧԺ��ѧ�о�����ޮ������������-�������̬���ת�Ʒ�Ӧ�о���ȡ����Ҫ��չ��ͼ��
��ޮ ��� ���ת�� ���� ��Ӧ����ѧ
2024/3/17
�����о�����̬���ӵķ�Ӧ����ѧ�������������Ǽʿռ䡢���Ǵ�������������ȸ������������������Ҫ��ѧ���塣Ar+ + N2 �� Ar + N2+���о����ת�Ʒ�Ӧ����ѧ�ľ���ģ����ϵ������ͬ��ʵ���о��Լ�ʵ������ۼ���֮����ںܶ����飬���Ƕ���һģ����ϵ�ĵ��ת�ƻ�����������Ȼ�dz����ޡ�

�й���ѧ������ѧ���˰����ڿ��������й���ѧԺ������ѧ�����о�����־���о�Ա����ѧ��Ժʿ����������������˻�Ԫ��ѧ��Ӧ����������ֲ������Ӹ�������ʾ�˵�������-�������öԻ�ѧ��Ӧ����ѧ���̵�Ӱ�졣��һ�о��ɹ���2021��2��26�շ����ڡ���ѧ����Science����־�ϡ�
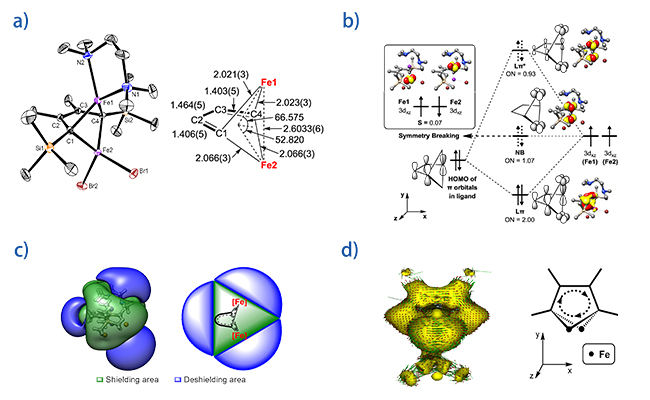
�й���ѧԺ������ѧ�����о�����ʾ��������ص��Ƿ�ƽ���������ķ�������Դ��ͼ��
������ص� ��ƽ���� ����� ��������Դ
2020/11/12
���գ����������������ص�ʵ����Ҷ�����о�Ա�Ŷ��뱱����ѧϯ�����ں������۽����о��ŶӺ������ɹ���ʾ�˷�ƽ�涡��ϩ��˫��������ķ�������Դ�����������ĵ�3dxz�붡��ϩ��֮��d-p����Ħ����ص����������ǻ�ѧ�Ļ�������֮һ���ڴ�������㻯�����У��乲�һ����ͨ�����֮��粢��Ħ��ͽ���ʵ�ֵ�������������Ǵ��������ƽ��ṹ�������ڽ����ӷ��㻯�����У����ڽ���d������в�ͬ����״�Ϳռ�ȡ��Ϊ...
ɽ��ʦ����ѧ�ṹ��ѧ�μ��ڶ��� ���ۼ����ۺͷ��ӽṹ2-4�ӻ��������
ɽ��ʦ����ѧ �ṹ��ѧ �μ� �ڶ��� ���ۼ����� ���ӽṹ �ӻ��������
2017/6/20
ɽ��ʦ����ѧ�ṹ��ѧ�μ��ڶ��� ���ۼ����ۺͷ��ӽṹ2-4�ӻ�������ۡ�
ɽ��ʦ����ѧ�ṹ��ѧ�μ���һ�� ������ѧ������ԭ�ӽṹ1-9�����ԭ�ӽṹ���۵��������ģ�͡�ԭ�����
ɽ��ʦ����ѧ �ṹ��ѧ �μ� ��һ�� ������ѧ���� ԭ�ӽṹ �����ԭ�ӽṹ���� �������ģ�� ԭ�ӹ��
2017/6/20
ɽ��ʦ����ѧ�ṹ��ѧ�μ���һ�� ������ѧ������ԭ�ӽṹ1-9�����ԭ�ӽṹ���۵��������ģ�͡�ԭ�������

���ڣ�ǿ�ų������Ų����о�Ա��������5dǿ����-�����ϲ���Sr2IrO4���о���ȡ�ý�չ����ع�����Enhanced electrical conductivity and diluted Ir4+ spin orders in electron doped iridates Sr2�CxGaxIrO4Ϊ�ⷢ���ڡ�Ӧ�������챨���ϡ�
��ѧ���ڵ�����������ͭ�����š�
ͭ������ �������ӻ�����
2015/5/21
2015��5��20�գ����ߴ��п�Ժ���ݻ�ѧ�����о�����Ϥ���ɸ����о�Ա�ƺ��������о�С�龭����4���Ŭ���������ɻ��ͼ��Ի�����֮�������һ��ͭ�ʵġ������š���ʵ�������ɻ��뼫�����ӻ�����֮��ĵ�����ת�ƣ����ҹ��������ɻ�������ת�Ƶ�����ż����Ӧ����سɹ���ǰ���߷����ڡ��¹�Ӧ�û�ѧ����־��
�����о���C5H��C5D���ɻ�A2��-X2�����������״�. ������Ȳ/�����������ֱ���ŵ������̼����������Ʊ�����C5H��C5D���ɻ������ù�ǻ˥������¼̼�������������ֱ�����չ���. ��֮ǰ������ȣ�ʵ����IJ���У�õ��˸Ľ�. ͨ����ʵ����ķ������״�ʵ��ȷ����C5H���ɻ�A2������̬������-������ѳ���Ϊ-0.7(3) cm-1. ��Ϊ��ʶA2�����̬���ṩ��������ʵ��֤��. ͨ����...
��������������3D-QSAR��N-�����������
N-�������� ����HIV-1���� ��ά������Ч��ϵ ���ӹ������ �ȽϷ��ӳ����� MOPAC6/PM3
2009/12/14
N-��������(NAIMs)��ͨ�����ֲ�ͬ�����÷�ʽ����HIV-1�ĸ���. �ñȽϷ��ӳ���CoMFA��������һϵ���й�ͬ�Ǽܵ�NAIM���ӽ���3D-QSARģ��. ������ģ�Ͳ�ͬ���ǣ���ƫ��С���ˣ�PLS�������г���������������������Ϣ���о���������������������Ĺ�ϵ. ����õ��˼���ģ�ͣ��������������ģ�͵Ĺ�����Ϊ21.7%�����HOMO5��ģ�͵Ĺ������.
����EHMO�������ĸ�[M_4S_2]��M��Co��Nb���ʹغ���ĵ��ӽṹ�����˼��㣬ͨ������ԭ��3d�������ǰ����ϵ������������ܼ�����ɷֲ���Mulliken���������ʵıȽϣ����ִغ���������������ԭ�ӵ�3d��������˳ɼ������ȶ�[M_4S_2]�ṹ��Ԫ����Ҫ�����ã�
����ģ�ͷ�����������������ж�����������Ȳ�Ħ�����������ã�Ӧ�ö���ɢ��X����Ǣ��������ģ�ͷ��ӵĵ��ӽṹ���м��㣬�õ��������ͨ���ռ�����õĴ�С�������������Ȳ���������ӳ�ָ���½����ڻ����������ӵ��ӽṹ�о������ϣ������˷������ͨ����������ã�����������������ͨ���ռ��������ͨ�������������������ù���̬�������㻷����������ǰ�߷�����������ܣ���ʵ����ϽϺ�
5,7��-(�Ǽװ���)-��-8-�ǻ������������л��������ܶȷ�������Ȼ������������ܶ����������о�
8-�ǻ���������� ��� ��Ȼ��������� �����ܶ����˷��� ��ʱ�ܶȷ��� ����
2009/11/25
�����ܶȷ�������, ��B3LYP/6-31Gˮƽ�϶�5,7��-(�Ǽװ���)-��-�ǻ��������3�ֽ���M(M=Zn, Mg, Be)�л������M(5,7��-Iminomethylq2)2�Ľṹ������ȫ�Ż�, ����TDDFT�������������չ���. ͬʱ, ������Ȼ���������(NBO)�͵����ܶ����˷���(AIM)�����Է�������������˷���. �������, ������ֵ��ʵ��ֵ��������, �����������нϴ��...
���ÿ��������ЧӦ��6-311G**ȫ���ӻ������ο�������, �����˸÷��ӵİ�������-������ЧӦ�Ĵ�ֱ�����ܺͻ�̬������̬C��I��������������. ���ۼ��㷢��, ��������33A''��11A'', 21A'���ֽ���, ����������C��I����Ϊ0.241 nm����; ��̬11A'������̬33A''(3Q0)�Ĵ�ֱ������Ϊ4.658 eV, ��ʵ��ֵ4.662 eV�dz��Ǻ�. ������C2F5I������...
�Ͽ���ѧ�ṹ��ѧ�μ������� ���ӽṹ������ϵ���ݿ˶�����������ۡ�
����DFT/B3LYP������ϵ�ж�����ϵ������ȫ�Ż�, ����ṹ�������жԱ�. �ڴ˻�����, �õ������ӵ����ռ���������Ϳ����������ϵ��HOMO-LUMO��϶, ����������϶�뵼���ԵĹ�ϵ��Ԥ�����������. �Ը����ӵ��������ѧ���ʽ������о�. ����ѧ�������������Ӿ����ȶ�, ���л�����DFBT���ȶ�. ����ZINDO��TD-DFT�������������չ���, �����ṹ�����Թ������ʵ�Ӱ��. ����...
�й��о����������а�-��
- ���ڼ���...
�й�ѧ���ڿ����а�-��
- ���ڼ���...
�����ѧ���л������а�-��
- ���ڼ���...
�й���ѧ���а�-��
- ���ڼ���...
�ˡ���-ƪ
- ���ڼ���...
�Ρ���-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
�������� -ƪ
- ���ڼ���...
֪ʶҪ��-ƪ
- ���ڼ���...
���ʶ�̬-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
ѧ��ָ��-ƪ
- ���ڼ���...
ѧ��վ��-ƪ
- ���ڼ���...